Alignment Gap Summary
gap.inspect.RdReport the number of gaps per sequence and per position for a given alignment.
gap.inspect(x)
Arguments
| x | a matrix or an alignment data structure obtained from
|
|---|
Details
Reports the number of gap characters per row (i.e. sequence) and
per column (i.e. position) for a given alignment. In addition,
the indices for gap and non-gap containing coloums are returned along
with a binary matrix indicating the location of gap positions.
Value
Returns a list object with the following components:
a numeric vector detailing the number of gaps per row (i.e. sequence).
a numeric vector detailing the number of gaps per column (i.e. position).
indices for gap containing coloums
indices for non-gap containing coloums
a binary numeric matrix with the same dimensions as the
alignment, with 0 at non-gap positions and 1 at gap
positions.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Author
Barry Grant
Note
During alignment, gaps are introduced into sequences that are believed to have undergone deletions or insertions with respect to other sequences in the alignment. These gaps, often referred to as indels, can be represented with ‘NA’, a ‘-’ or ‘.’ character.
This function gives an overview of gap occurrence and may be useful when considering positions or sequences that could/should be excluded from further analysis.
See also
read.fasta, read.fasta.pdb
Examples
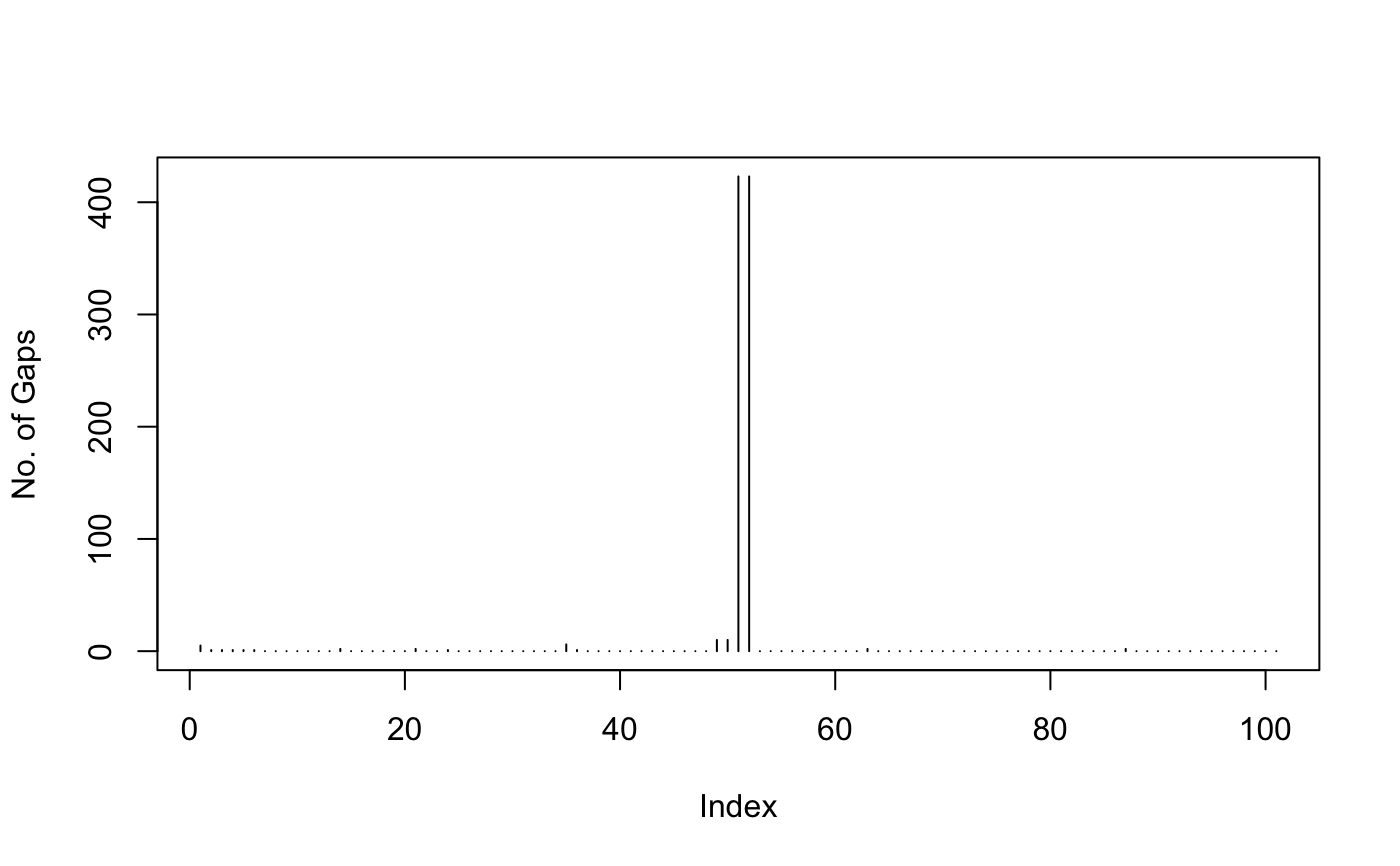
aln <- read.fasta( system.file("examples/hivp_xray.fa", package = "bio3d") ) gap.stats <- gap.inspect(aln$ali) gap.stats$row # Gaps per sequence#> [1] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 0 0 2 2 2 2 2 2 2 2 #> [38] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 4 3 2 2 4 #> [75] 3 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 3 2 2 2 2 2 2 #> [112] 6 6 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 4 4 2 2 2 2 2 2 2 #> [149] 2 2 2 2 2 6 5 4 4 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 #> [186] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 2 2 2 2 2 2 2 2 2 2 2 2 2 4 5 2 2 2 #> [223] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 #> [260] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 #> [297] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 3 3 2 8 2 #> [334] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 #> [371] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 #> [408] 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2gap.stats$col # Gaps per position#> [1] 5 1 1 1 1 1 0 0 0 0 0 0 0 2 0 0 0 0 #> [19] 0 0 2 0 0 1 0 0 0 0 0 0 0 0 0 0 6 1 #> [37] 0 0 0 0 0 0 0 0 0 0 0 0 10 10 423 423 0 0 #> [55] 0 0 0 0 0 0 0 0 2 0 0 0 0 0 0 0 0 0 #> [73] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 2 0 0 0 #> [91] 0 0 0 0 0 0 0 0 0 0 0##gap.stats$bin # Binary matrix (1 for gap, 0 for aminoacid) ##aln[,gap.stats$f.inds] # Alignment without gap positions plot(gap.stats$col, typ="h", ylab="No. of Gaps")