Beginning Trajectory Analysis with Bio3D
Xin-Qiu Yao, Lars Skjaerven, & Barry J. Grant
Bio3D_md.RmdBackground
Bio3D1 is a group of R packages that provide interactive tools for the analysis of bimolecular structure, sequence and simulation data. The aim of this document, termed a vignette2 in R parlance, is to provide a brief task-oriented introduction to basic molecular dynamics trajectory analysis with the Bio3D core package, bio3d (Grant et al. 2006).
Requirements
Detailed instructions for obtaining and installing the Bio3D packages on various platforms can be found in the Installing Bio3D vignette available online. This particular vignette was generated using bio3d version 2.4.1.9000.
Getting Started
Start R, load the bio3d package and use the command demo("md") to get a quick feel for some of the tasks that we will be introducing in the following sections.
Side-note:
Note that you will be prompted to hit the RETURN key at each step of the demo as this will allow you to see the particular functions being called. Also note that detailed documentation and example code for each function can be accessed via the help() and example() commands (e.g. help(read.pdb)). You can also copy and paste any of the example code from the documentation of a particular function, or indeed this vignette, directly into your R session to see how things work. You can also find this documentation online.
Reading Example Trajectory Data
A number of example data sets are shipped with the bio3d package. The main purpose of including this data is to allow users to more quickly appreciate the capabilities of various Bio3D functions that would otherwise require potentially time consuming data generation. In the examples below we will input, process and analyze a molecular dynamics trajectory of Human Immunodeficiency Virus aspartic protease (HIVpr). This trajectory is stored in CHARMM/NAMD DCD format and has had all solvent and non C-alpha protein atoms excluded to reduce overall file size.
The code snippet below sets the file paths for the example HIVpr starting structure (pdbfile) and trajectory data (dcdfile).
dcdfile <- system.file("examples/hivp.dcd", package="bio3d") pdbfile <- system.file("examples/hivp.pdb", package="bio3d")
Side-note:
Note that in the above example the system.file() command returns a character string corresponding to the file name of a PDB structure included with the bio3d package. This is required as users may install the package in different locations. When using your own input files the system.file() command will not be required, for example
mydcdfile <- "/path/to/my/data/myfile.dcd"
The read.dcd() and read.pdb() commands processes the input files and returns their output to the new objects dcd and pdb. We can check the basic structure of these objects with the following commands:
print(pdb)
##
## Call: read.pdb(file = pdbfile)
##
## Total Models#: 1
## Total Atoms#: 198, XYZs#: 594 Chains#: 2 (values: A B)
##
## Protein Atoms#: 198 (residues/Calpha atoms#: 198)
## Nucleic acid Atoms#: 0 (residues/phosphate atoms#: 0)
##
## Non-protein/nucleic Atoms#: 0 (residues: 0)
## Non-protein/nucleic resid values: [ none ]
##
## Protein sequence:
## PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD
## QILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFPQITLWQRPLVTIKIGGQLKE
## ALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTP
## VNIIGRNLLTQIGCTLNF
##
## + attr: atom, xyz, helix, sheet, calpha,
## callprint(pdb$xyz)
##
## Total Frames#: 1
## Total XYZs#: 594, (Atoms#: 198)
##
## [1] 51.842 59.784 -6.815 <...> 55.089 55.232 -6.899 [594]
##
## + attr: Matrix DIM = 1 x 594print(dcd)
##
## Total Frames#: 117
## Total XYZs#: 594, (Atoms#: 198)
##
## [1] 51.842 59.784 -6.815 <...> 27.995 70.42 -33.908 [69498]
##
## + attr: Matrix DIM = 117 x 594The output of the above function is telling us that we have 351 trajectory frames and 594 coordinates. The trajectory information is stored in a xyz matrix object with one row per frame and columns for the Cartesian coordinates (x, y and z columns).
Question:
How would you extract the amino acid sequence of the HIVpr system in 1-letter and 3-letter forms? HINT: try help.search("PDB sequence") for a Bio3D function that might help you.
Side-note:
Note that typically one works with trajectory files that contain all protein atoms, or at the very least all backbone atoms. Solvent however can often be excluded prior to Bio3D input - it just depends upon your particular analysis questions. For example, we are not able to analyze hydrogen bonding patterns or details of water occupancy with the currently inputed data.
Trajectory Frame Superposition
In this simple example we select all C-alpha atoms for trajectory frame superposition.
ca.inds <- atom.select(pdb, elety="CA")
The returned ca.inds object is a list containing atom and xyz numeric indices that we can now use to superpose all frames of the trajectory on the selected indices (in this case corresponding to all alpha Carbon atoms). For this we will with the fit.xyz() function.
xyz <- fit.xyz(fixed=pdb$xyz, mobile=dcd, fixed.inds=ca.inds$xyz, mobile.inds=ca.inds$xyz)
The above command performs the actual superposition and stores the new coordinates in the matrix object xyz. Note that the dimensions (i.e. number of rows and columns, which correspond to frames and coordinates respectively) of xyz match those of the input trajectory:
## [1] TRUE TRUEQuestion:
How would you fit trajectory frames on the Calpha atoms of residues 24 to 27 and 85 to 90 in both chains? HINT: See the example section of help(atom.select).
Question:
Would you expect the alternate fitting suggested above to alter your later results? HINT: You can come back to this question later after going through the other sections.
Side-note:
A simple way to obtain the average structure from your fitted trajectory is to use the following command colMeans(xyz).
Root Mean Square Deviation (RMSD)
RMSD is a standard measure of structural distance between coordinate sets and is implemented in the Bio3D function rmsd().
rd <- rmsd(xyz[1,ca.inds$xyz], xyz[,ca.inds$xyz]) plot(rd, typ="l", ylab="RMSD", xlab="Frame No.") points(lowess(rd), typ="l", col="red", lty=2, lwd=2)
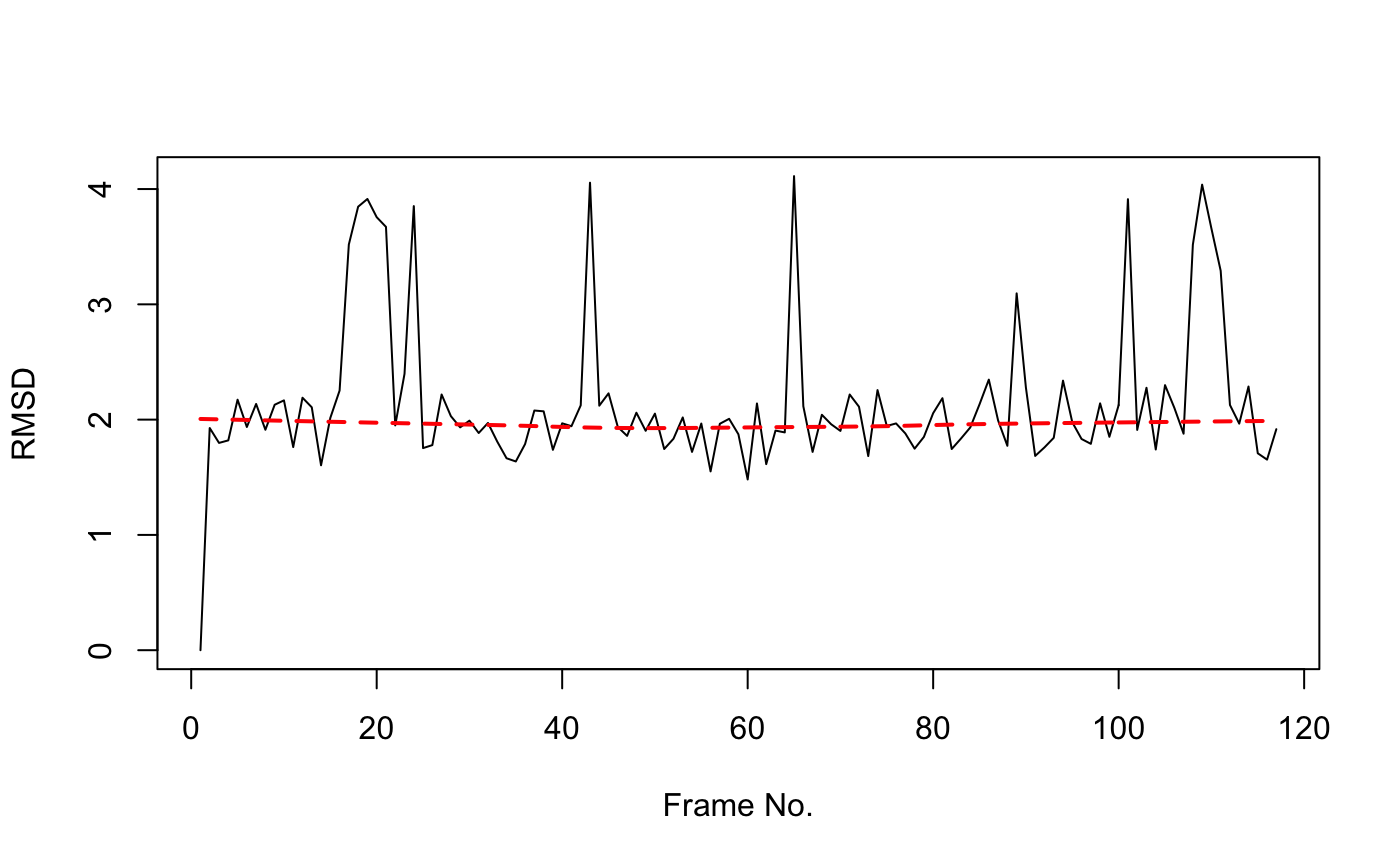
Simple time series of RMSD from the initial structure (note periodic jumps that we will later see correspond to transient openings of the flap regions of HIVpr)
A quick histogram can be useful for examining the distribution of RMSD values.
hist(rd, breaks=40, freq=FALSE, main="RMSD Histogram", xlab="RMSD") lines(density(rd), col="gray", lwd=3)
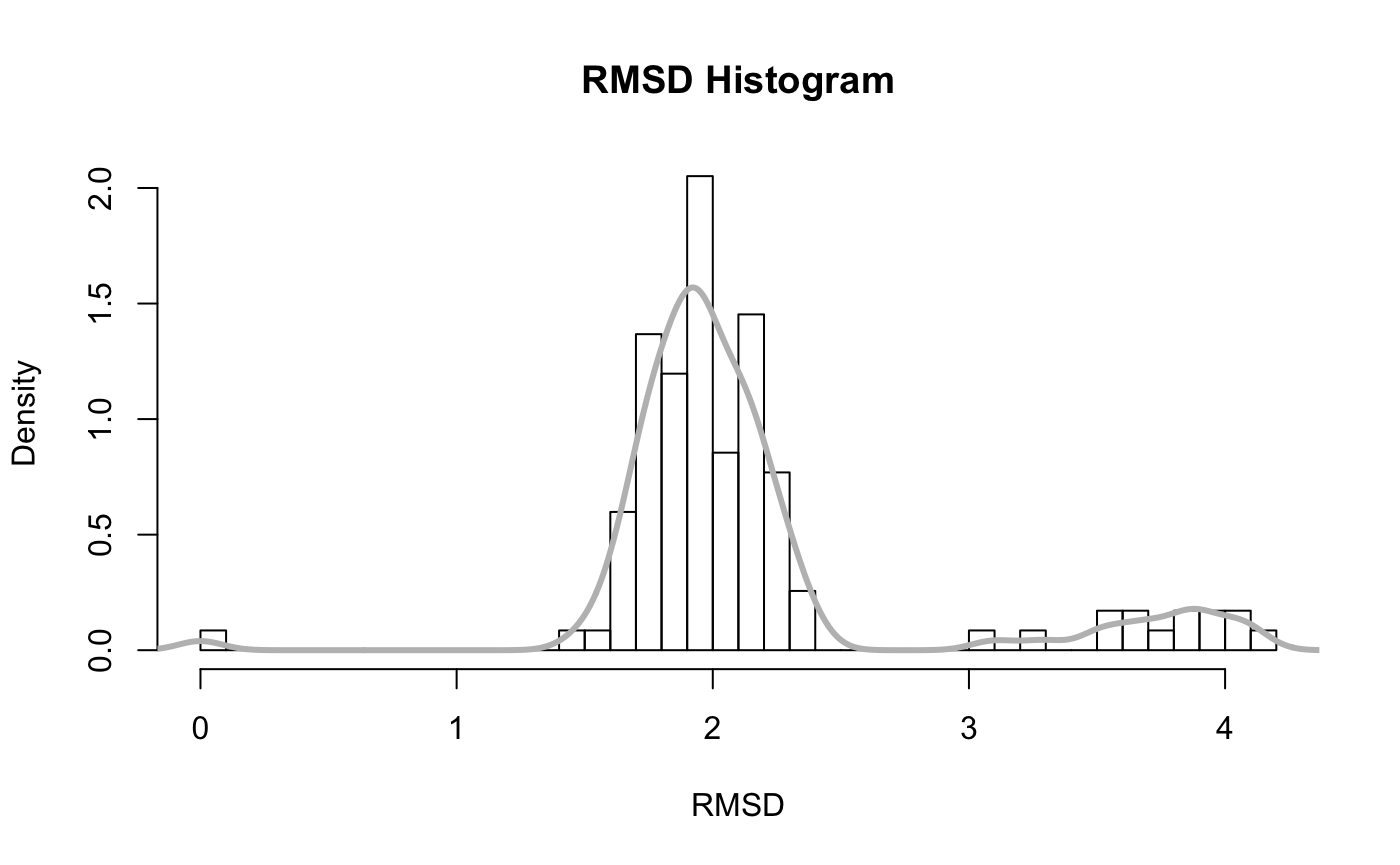
Note the spread of RMSD values and that the majority of sampled conformations are around 2 Angstroms from the starting structure
summary(rd)
## Min. 1st Qu. Median Mean 3rd Qu. Max.
## 0.000 1.834 1.967 2.151 2.167 4.112Root Mean Squared Fluctuations (RMSF)
RMSF is an often used measure of conformational variance and is implemented in the Bio3D function rmsf(). This analysis will highlight the portions of structure that are fluctuating from their mean structure the most (and least).
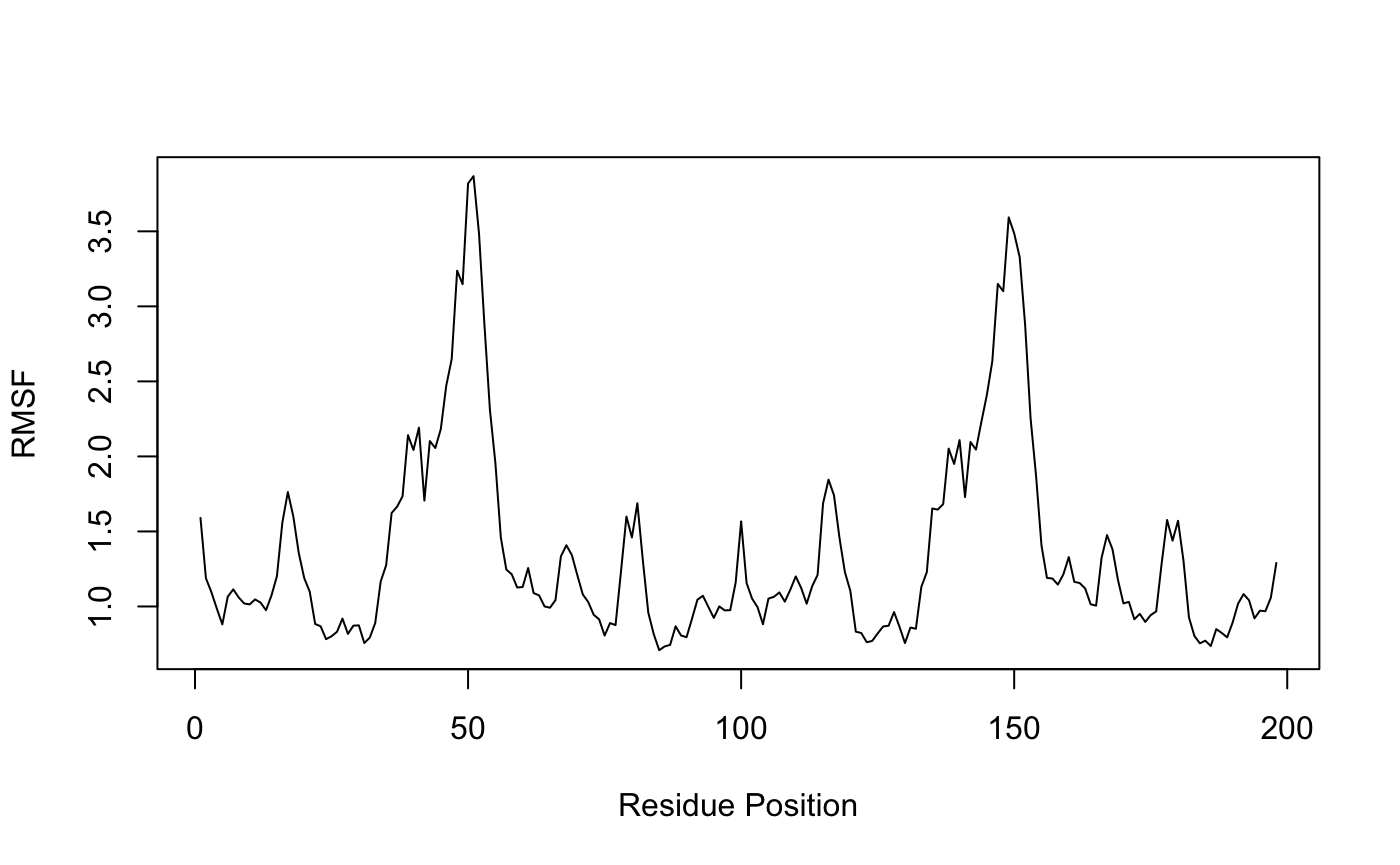
Residue-wise RMSF indicates regions of high mobility
Question:
If you had multiple simulations (which by the way we greatly encourage), how would you plot these on the same graph? HINT: See help(points).
Question:
If you noticed differences at particular sites (e.g. in the presence or absence of a ligand) how would you go about addressing the significance of these differences? HINT: this is why we always encourage a multiple simulation approach and implement Bio3D in one of the most advanced statistical analysis packages available.
Principal Component Analysis
PCA can be employed to examine the relationship between different conformations sampled during the trajectory and is implemented in the Bio3D functions pca.xyz() and pca.tor(). The application of PCA to both distributions of experimental structures and molecular dynamics trajectories will be covered in detail in other vignettes. Briefly, we will note here that this method can provide considerable insight into the nature of conformational differences with the resulting principal components (orthogonal eigenvectors) describing the axes of maximal variance of the distribution of structures. Projection of the distribution onto the subspace defined by the largest principal components results in a lower dimensional representation of the structural dataset (see Figure 4). The percentage of the total mean square displacement (or variance) of atom positional fluctuations captured in each dimension is characterized by their corresponding eigenvalue (see Figure 4D). Experience suggests that 3–5 dimensions are often sufficient to capture over 70 percent of the total variance in a given family of experimental structures or indeed a standard molecular dynamics trajectory. Thus, a handful of principal components are sufficient to provide a useful description while still retaining most of the variance in the original distribution (Grant et al. 2006).
A quick overview of the results of pca.xyz() can be obtained by calling plot.pca()
pc <- pca.xyz(xyz[,ca.inds$xyz]) plot(pc, col=bwr.colors(nrow(xyz)) )
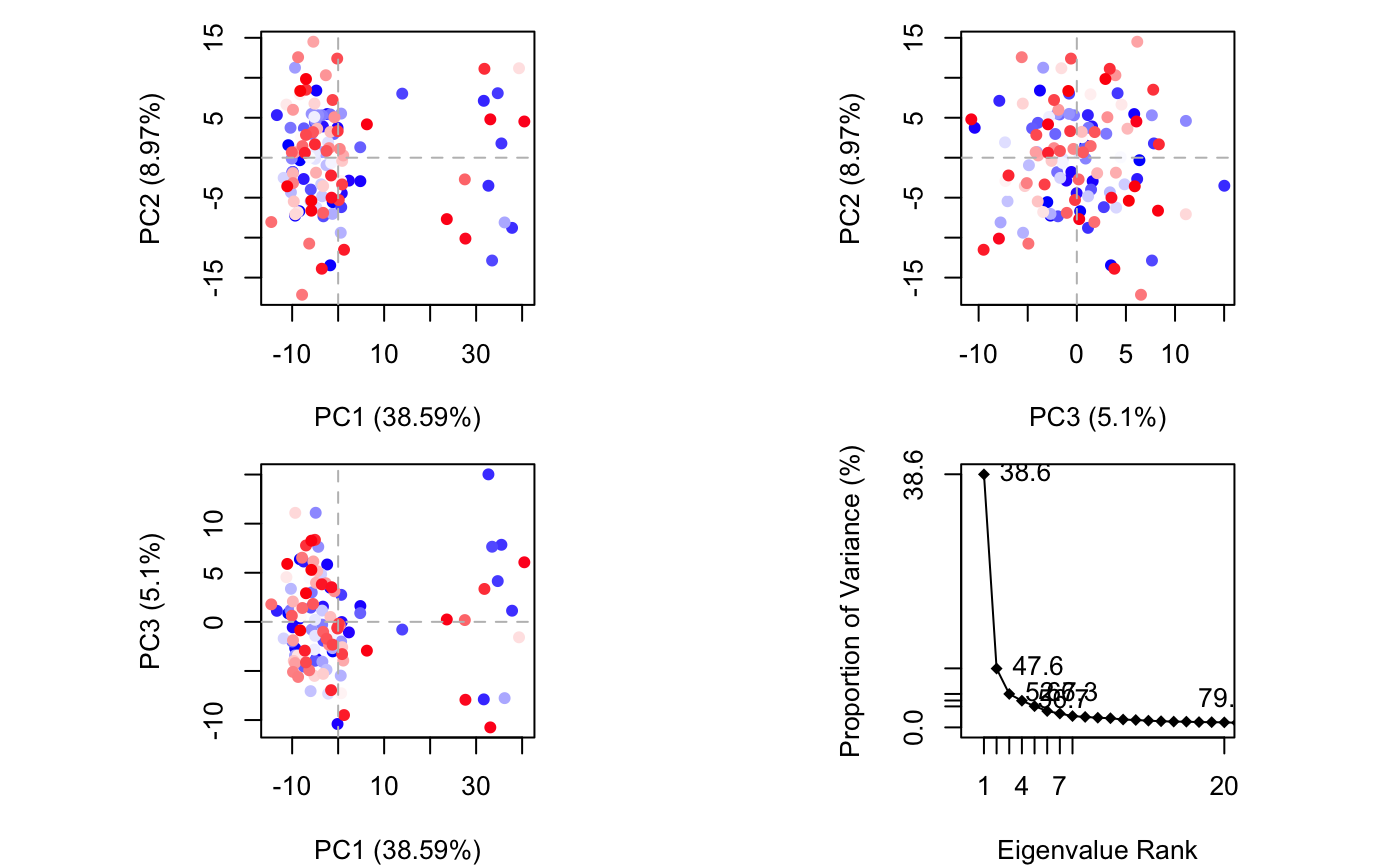
PCA results for our HIVpr trajectory with instantaneous conformations (i.e. trajectory frames) colored from blue to red in order of time
Note that there are distinct groupings of conformations along the PC1 plane (one centered around +30 and a second, larger grouping, at -5). The continuous color scale (from blue to whit to red) indicates that there are periodic jumps between these conformers throughout the trajectory. Below we perform a quick clustering in PC-space to further highlight these distinct conformers.
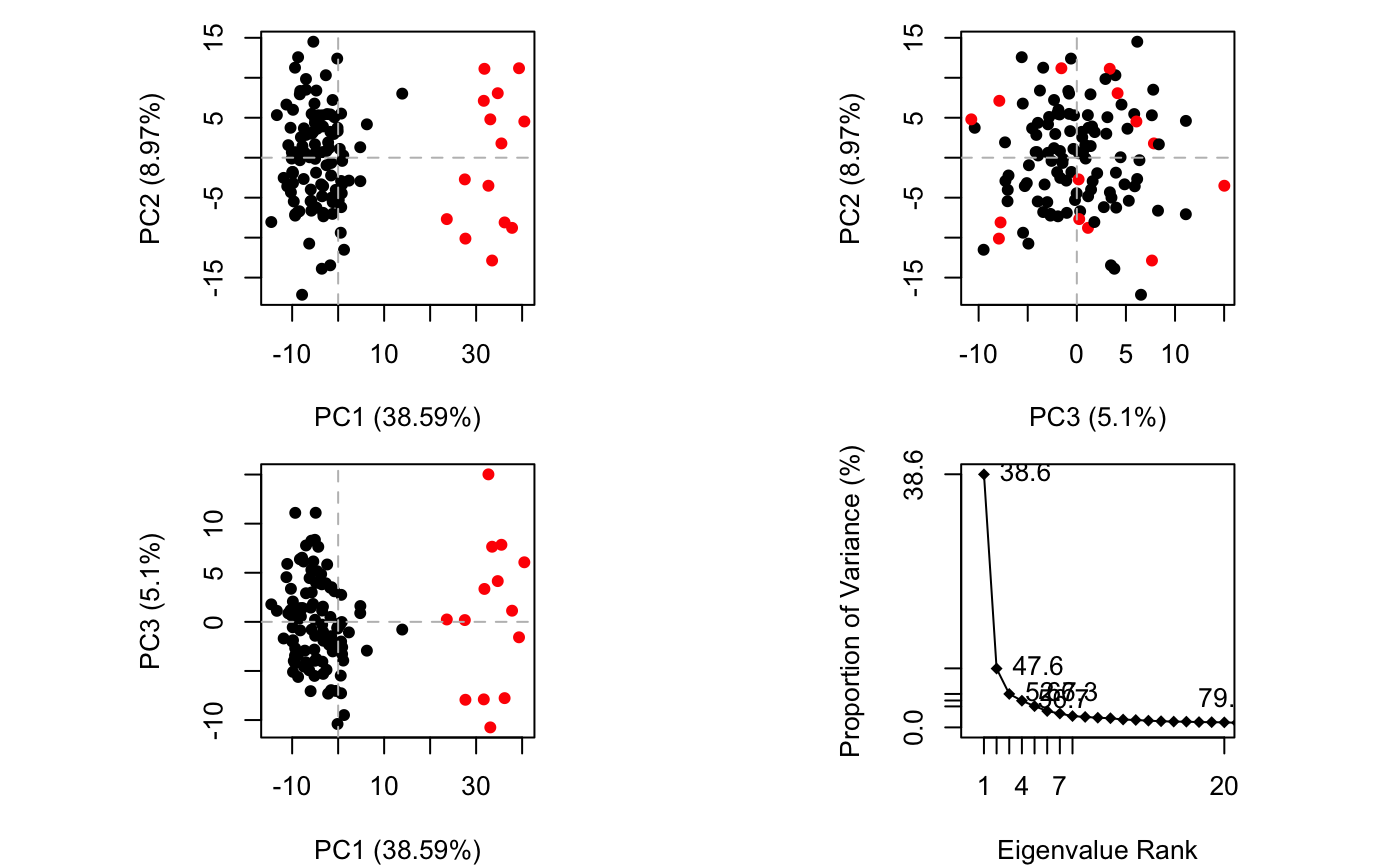
Simple clustering in PC subspace
Question:
How can we find out which frames correspond to the distinct groups along PC1? HINT: Which variable/object created above tells us about cluster membership?
Bellow we call plot.bio3d() to examine the contribution of each residue to the first two principal components.
plot.bio3d(pc$au[,1], ylab="PC1 (A)", xlab="Residue Position", typ="l") points(pc$au[,2], typ="l", col="blue")
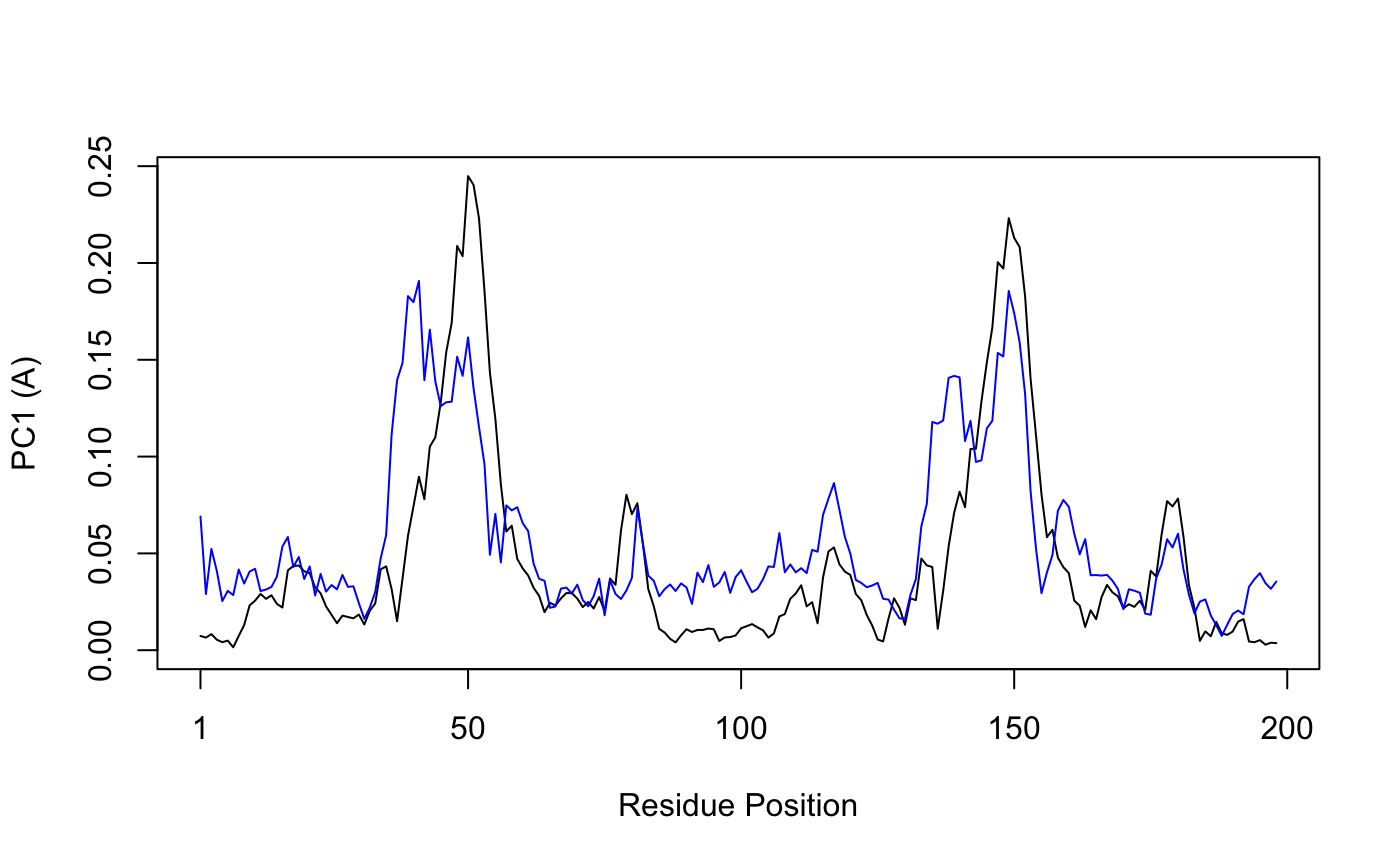
Residue-wise loadings for PC1 (black) and PC2 (blue)
Question:
Why do you think there might be two major peaks in our RMSF plot?
To further aid interpretation, a PDB format trajectory can be produced that interpolates between the most dissimilar structures in the distribution along a given principal component. This involves dividing the difference between the conformers into a number of evenly spaced steps along the principal components, forming the frames of the output multi-model PDB trajectory. Such trajectories can be directly visualized in a molecular graphics program, such as VMD (Humphrey 1996). Furthermore, the interpolated structures can be analyzed for possible domain and shear movements with other Bio3D functions, or used as initial seed structures for reaction path refinement methods (note you will likely want to perform all heavy atom PCA for such applications).
p1 <- mktrj.pca(pc, pc=1, b=pc$au[,1], file="pc1.pdb") p2 <- mktrj.pca(pc, pc=2, b=pc$au[,2], file="pc2.pdb")
You can also write these trajectory’s as AMBER NetCDF format files with the write.ncdf function. To view the PDB trajectories in VMD just open the files in the normal way and display as tube representation for example (see figure below).
write.ncdf(p1, "trj_pc1.nc")
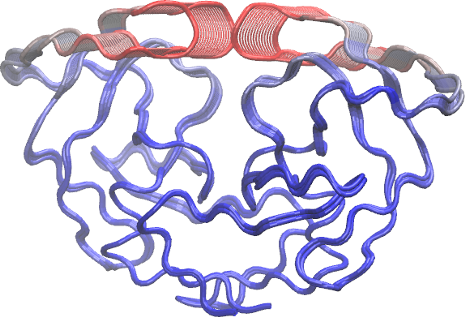
Visualization of the first principal component of HIV-1 protease. Color scale from blue to red depict low to high atomic displacements.
Cross-Correlation Analysis
The extent to which the atomic fluctuations/displacements of a system are correlated with one another can be assessed by examining the magnitude of all pairwise cross-correlation coefficients. The Bio3D dccm() function returns a matrix of all atom-wise cross-correlations whose elements may be displayed in a graphical representation frequently termed a dynamical cross-correlation map, or DCCM.
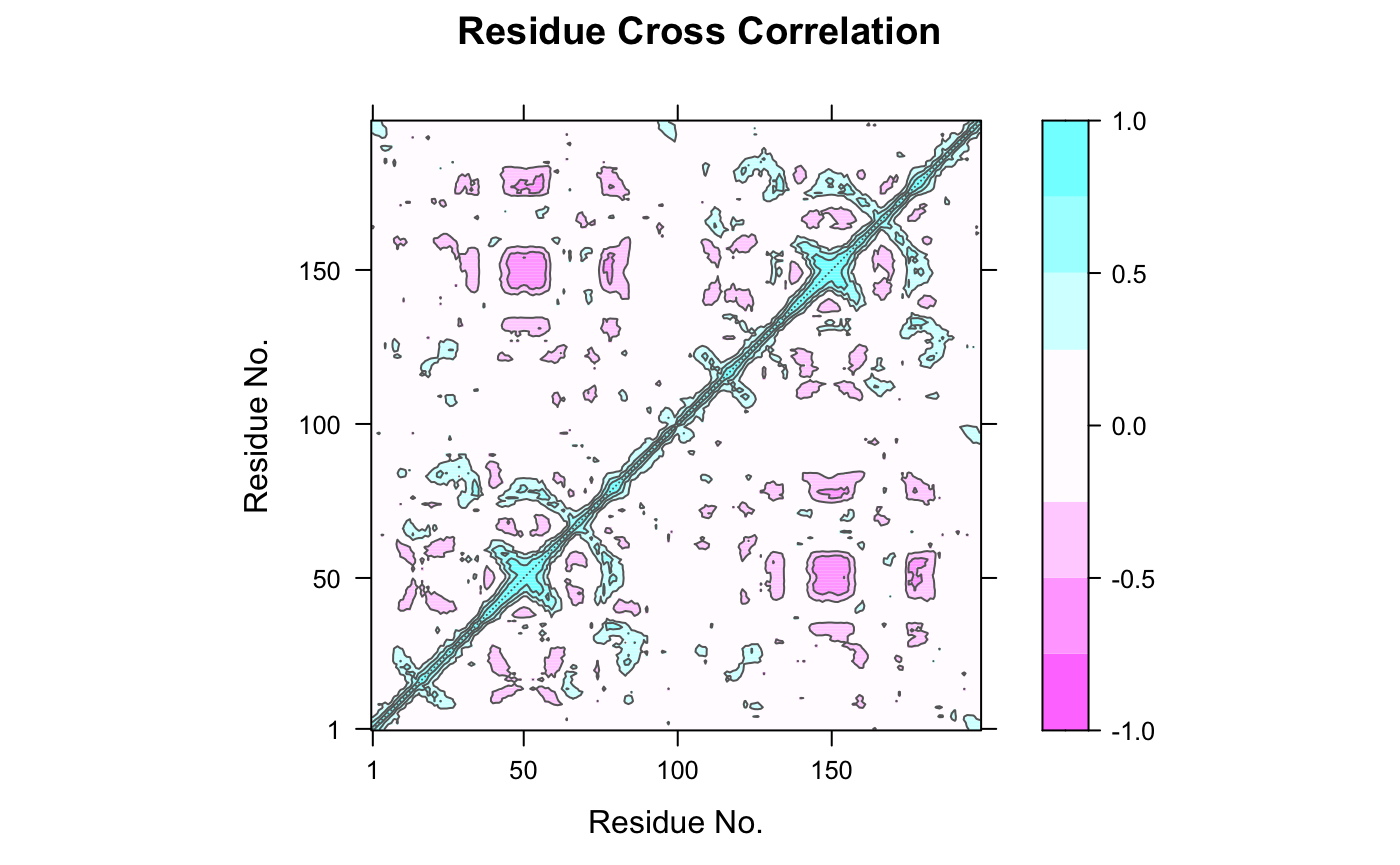
Dynamical cross-correlation map (DCCM) calculated with function dccm() using the MD simulation of HIV-1 protease as input.
Question:
What do the off-diagonal regions of negative correlation correspond to in this plot and which regions are involved? HINT: Negative values typical indicate regions that move in opposite directions.
A 3D visualization of these correlations can be provided through the function pymol.dccm()
# View the correlations in pymol pymol.dccm(cij, pdb, type="launch")
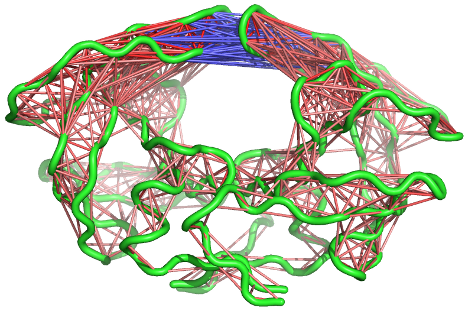
Visualization of residue-residue cross correlations (using function pymol.dccm()). Red and blue lines indicate correlated and anti-correlation motions, respectively.
See also the Enhanced Methods for Normal Mode Analysis for additional visualization examples. Also you might want to checkout the Comparative Analysis of Protein Structures vignette for relating results like these to available experimental data. The logical expansion of this analysis is described in the Correlation Network Analysis vignette.
Where to Next
If you have read this far, congratulations! We are ready to have some fun and move to other package vignettes that describe more interesting analysis including Correlation Network Analysis (where we will build and dissect dynamic networks form different correlated motion data), enhanced methods for Normal Mode Analysis (where we will explore the dynamics of large protein families and superfamilies), and advanced Comparative Structure Analysis (where we will mine available experimental data and supplement it with simulation results to map the conformational dynamics and coupled motions of proteins).
Document Details
This document is available from the Bio3D website in R markdown, HTML, and PDF formats. All code can be extracted and automatically executed to generate Figures and/or the PDF with the following commands:
Information About the Current Bio3D Session
## R version 3.6.0 (2019-04-26)
## Platform: x86_64-apple-darwin15.6.0 (64-bit)
## Running under: macOS 10.15.5
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/3.6/Resources/lib/libRlapack.dylib
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] bio3d_2.4-1.9000
##
## loaded via a namespace (and not attached):
## [1] Rcpp_1.0.5 lattice_0.20-41 digest_0.6.25 crayon_1.3.4
## [5] rprojroot_1.3-2 assertthat_0.2.1 grid_3.6.0 R6_2.4.1
## [9] backports_1.1.8 magrittr_1.5 evaluate_0.14 highr_0.8
## [13] stringi_1.4.6 rlang_0.4.6.9000 fs_1.4.2 rmarkdown_2.3.2
## [17] pkgdown_1.5.1.9000 desc_1.2.0 tools_3.6.0 stringr_1.4.0
## [21] parallel_3.6.0 yaml_2.2.1 xfun_0.15 compiler_3.6.0
## [25] memoise_1.1.0 htmltools_0.5.0 knitr_1.29References
Grant, B. J., A. P. D. C Rodrigues, K. M. Elsawy, A. J. Mccammon, and L. S. D. Caves. 2006. “Bio3d: An R Package for the Comparative Analysis of Protein Structures.” Bioinformatics 22: 2695–6.
Humphrey, et al., W. 1996. “VMD: Visual Molecular Dynamics.” J. Mol. Graph 14: 33–38.
The latest version of the package, full documentation and further vignettes (including detailed installation instructions) can be obtained from the main Bio3D website: http://thegrantlab.org/bio3d/↩︎
This vignette contains executable examples, see
help(vignette)for further details.↩︎