Principal Component Analysis
pca.xyz.RdPerforms principal components analysis (PCA) on a xyz
numeric data matrix.
# S3 method for xyz pca(xyz, subset = rep(TRUE, nrow(as.matrix(xyz))), use.svd = FALSE, rm.gaps=FALSE, mass = NULL, ...) # S3 method for pca print(x, nmodes=6, ...)
Arguments
| xyz | numeric matrix of Cartesian coordinates with a row per structure. |
|---|---|
| subset | an optional vector of numeric indices that selects a
subset of rows (e.g. experimental structures vs molecular dynamics
trajectory structures) from the full |
| use.svd | logical, if TRUE singular value decomposition (SVD) is called instead of eigenvalue decomposition. |
| rm.gaps | logical, if TRUE gap positions (with missing coordinate data in any input structure) are removed before calculation. This is equivalent to removing NA cols from xyz. |
| x | an object of class |
| nmodes | numeric, number of modes to be printed. |
| mass | a ‘pdb’ object or numeric vector of residue/atom masses.
By default ( |
| ... | additional arguments to |
Note
If mass is provided, mass weighted coordinates will be considered,
and iteration of fitting onto the mean structure is performed internally.
The extra fitting process is to remove external translation and rotation
of the whole system. With this option, a direct comparison can be made
between PCs from pca.xyz and vibrational modes from
nma.pdb, with the fact that
$$A=k_BTF^{-1}$$,
where \(A\) is the variance-covariance matrix, \(F\) the Hessian
matrix, \(k_B\) the Boltzmann's constant, and \(T\) the
temperature.
Value
Returns a list with the following components:
eigenvalues.
eigenvectors (i.e. the x, y, and z variable loadings).
scores of the supplied xyz on the pcs.
atom-wise loadings (i.e. xyz normalized eigenvectors).
the standard deviations of the pcs.
the means that were subtracted.
References
Grant, B.J. et al. (2006) Bioinformatics 22, 2695--2696.
Author
Barry Grant
See also
pca, pca.pdbs,
plot.pca, mktrj.pca,
pca.tor, project.pca
Examples
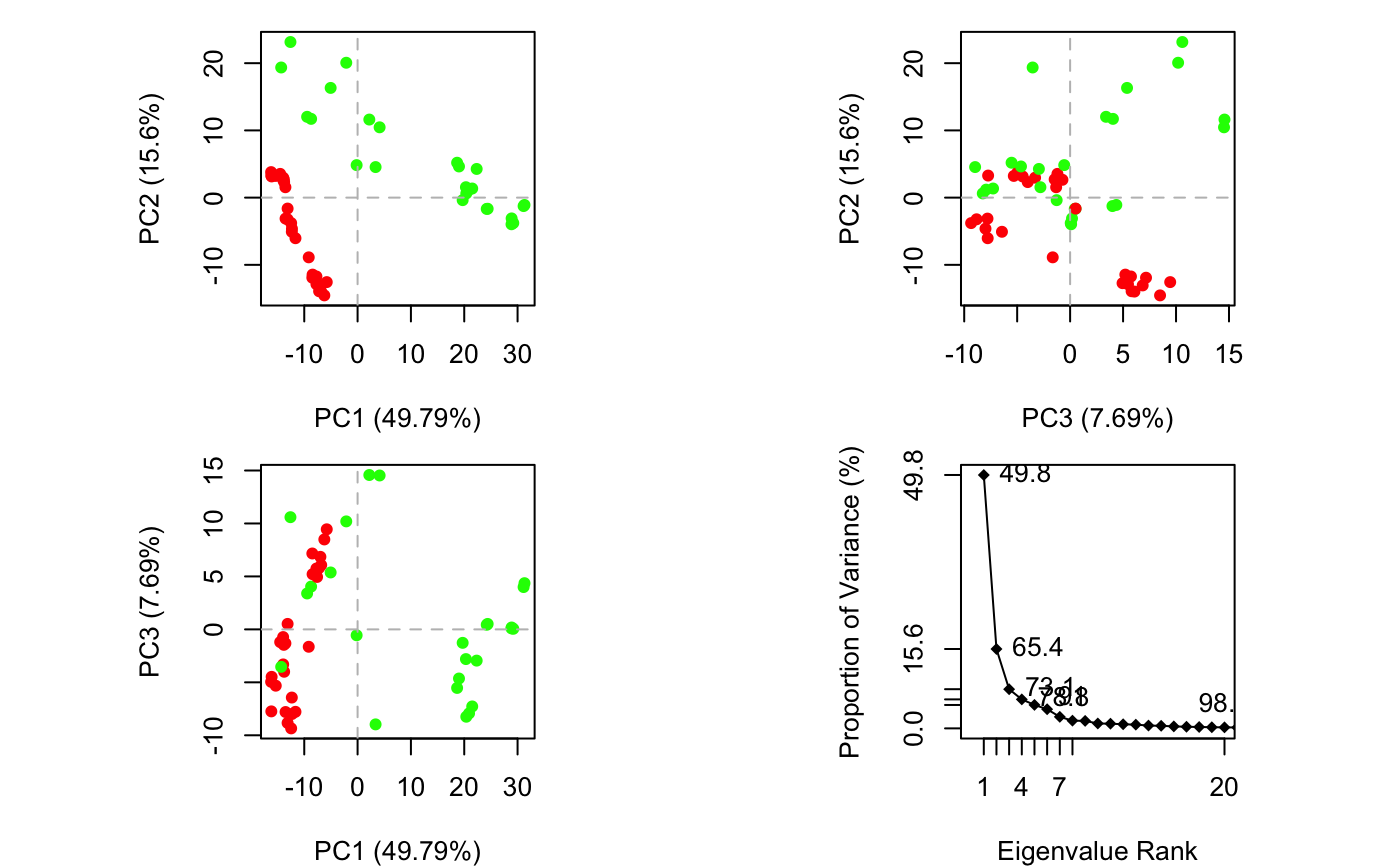
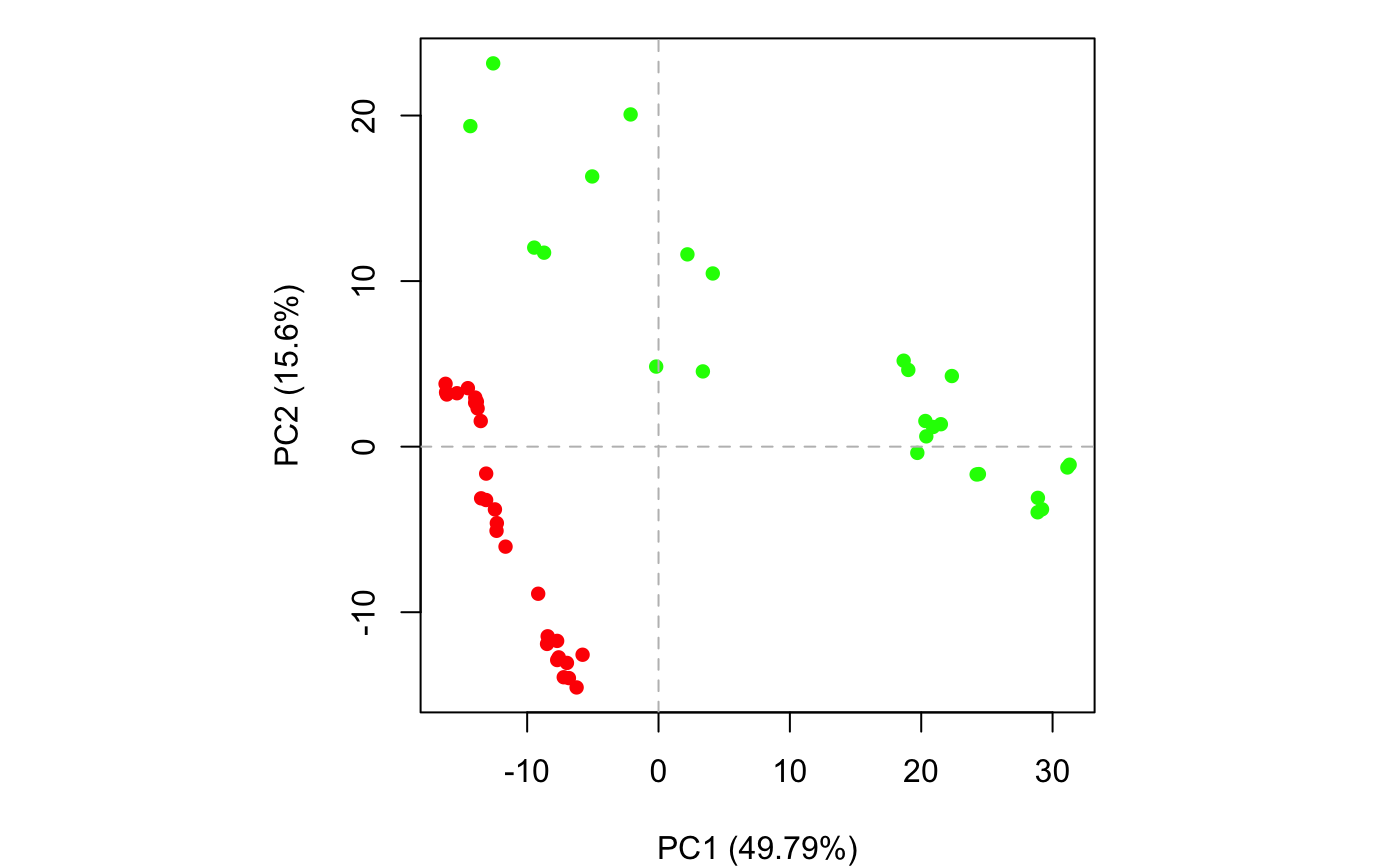
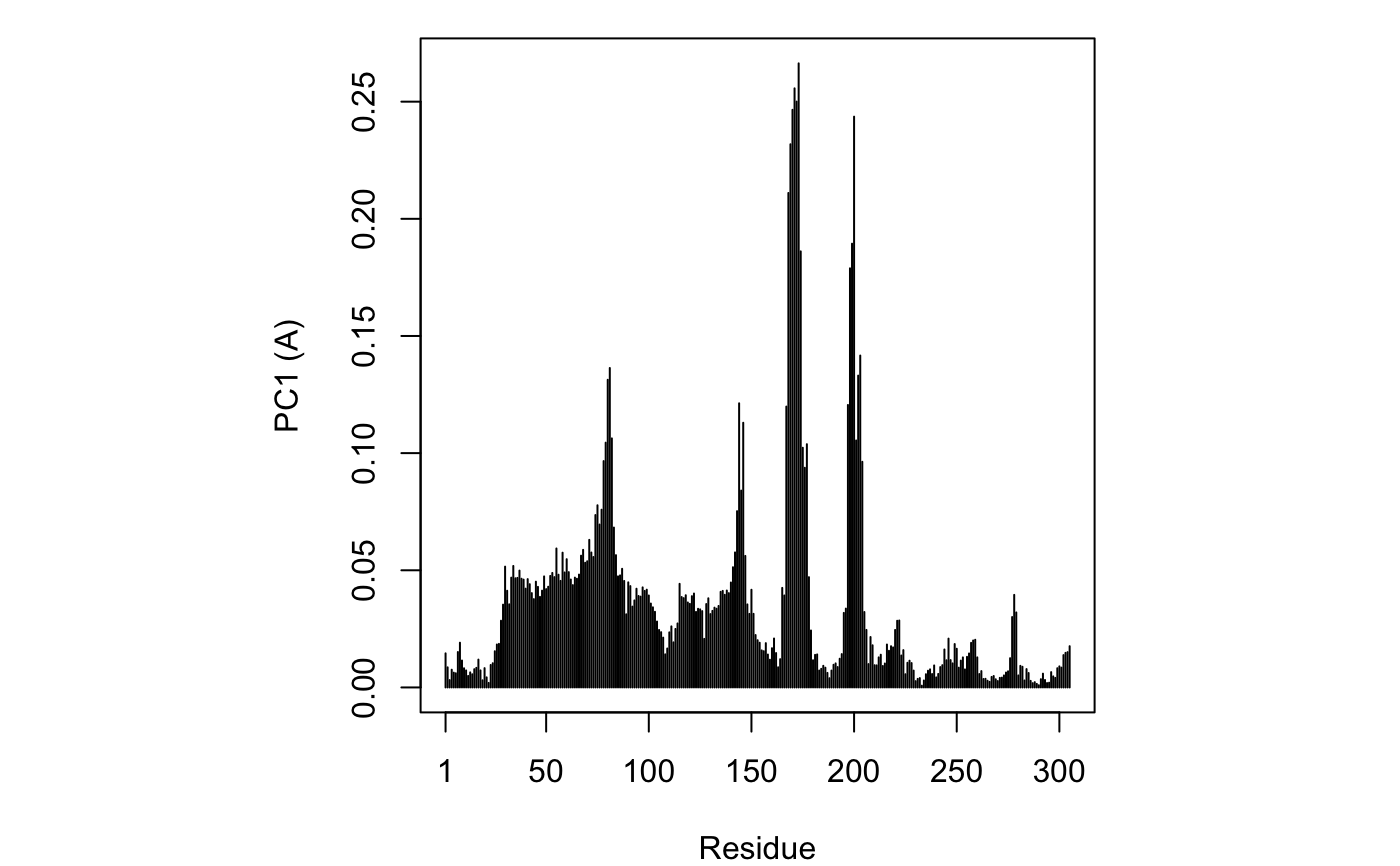
if (FALSE) { #-- Read transducin alignment and structures aln <- read.fasta(system.file("examples/transducin.fa",package="bio3d")) pdbs <- read.fasta.pdb(aln) # Find core core <- core.find(pdbs, #write.pdbs = TRUE, verbose=TRUE) rm(list=c("pdbs", "core")) } #-- OR for demo purposes just read previously saved transducin data attach(transducin) # Previously fitted coordinates based on sub 1.0A^3 core. See core.find() function. xyz <- pdbs$xyz #-- Do PCA ignoring gap containing positions pc.xray <- pca.xyz(xyz, rm.gaps=TRUE)#> NOTE: Removing 49 gap positions with missing coordinate data #> retaining 305 non-gap positions for analysis.## Plot atom wise loadings plot.bio3d(pc.xray$au[,1], ylab="PC1 (A)")# \donttest{ # PDB server connection required - testing excluded ## Plot loadings in relation to reference structure 1TAG pdb <- read.pdb("1tag")#> Note: Accessing on-line PDB file#> Warning: /var/folders/xf/qznxnpf91vb1wm4xwgnbt0xr0000gn/T//Rtmp4WslmZ/1tag.pdb exists. Skipping downloadind <- grep("1TAG", pdbs$id) ## location in alignment resno <- pdbs$resno[ind, !is.gap(pdbs)] ## non-gap residues tpdb <- trim.pdb(pdb, resno=resno) op <- par(no.readonly=TRUE) par(mfrow = c(3, 1), cex = 0.6, mar = c(3, 4, 1, 1)) plot.bio3d(pc.xray$au[,1], resno, ylab="PC1 (A)", sse=tpdb) plot.bio3d(pc.xray$au[,2], resno, ylab="PC2 (A)", sse=tpdb) plot.bio3d(pc.xray$au[,3], resno, ylab="PC3 (A)", sse=tpdb)par(op) # } if (FALSE) { # Write PC trajectory resno = pdbs$resno[1, !is.gap(pdbs)] resid = aa123(pdbs$ali[1, !is.gap(pdbs)]) a <- mktrj.pca(pc.xray, pc=1, file="pc1.pdb", resno=resno, resid=resid ) b <- mktrj.pca(pc.xray, pc=2, file="pc2.pdb", resno=resno, resid=resid ) c <- mktrj.pca(pc.xray, pc=3, file="pc3.pdb", resno=resno, resid=resid ) } detach(transducin)